- Page 1
- Page 2
- Page 3
- Page 4
- Page 5
- Page 6
- Page 7
- Page 8
- Page 9
- Page 10
- Page 11
- Page 12
- Page 13
- Page 14
- Page 15
- Page 16
- Page 17
- Page 18
- Page 19
- Page 20
- Page 21
- Page 22
- Page 23
- Page 24
- Page 25
- Page 26
- Page 27
- Page 28
- Page 29
- Page 30
- Page 31
- Page 32
- Page 33
- Page 34
- Page 35
- Page 36
- Page 37
- Page 38
- Page 39
- Page 40
- Page 41
- Page 42
- Page 43
- Page 44
- Page 45
- Page 46
- Page 47
- Page 48
- Page 49
- Page 50
- Page 51
- Page 52
- Page 53
- Page 54
- Page 55
- Page 56
- Page 57
- Page 58
- Page 59
- Page 60
- Page 61
- Page 62
- Page 63
- Page 64
- Page 65
- Page 66
- Page 67
- Page 68
- Page 69
- Page 70
- Flash version
© UniFlip.com
- Page 2
- Page 3
- Page 4
- Page 5
- Page 6
- Page 7
- Page 8
- Page 9
- Page 10
- Page 11
- Page 12
- Page 13
- Page 14
- Page 15
- Page 16
- Page 17
- Page 18
- Page 19
- Page 20
- Page 21
- Page 22
- Page 23
- Page 24
- Page 25
- Page 26
- Page 27
- Page 28
- Page 29
- Page 30
- Page 31
- Page 32
- Page 33
- Page 34
- Page 35
- Page 36
- Page 37
- Page 38
- Page 39
- Page 40
- Page 41
- Page 42
- Page 43
- Page 44
- Page 45
- Page 46
- Page 47
- Page 48
- Page 49
- Page 50
- Page 51
- Page 52
- Page 53
- Page 54
- Page 55
- Page 56
- Page 57
- Page 58
- Page 59
- Page 60
- Page 61
- Page 62
- Page 63
- Page 64
- Page 65
- Page 66
- Page 67
- Page 68
- Page 69
- Page 70
- Flash version
© UniFlip.com
Böbreğin Kistik Hastalıkları 575
A
B
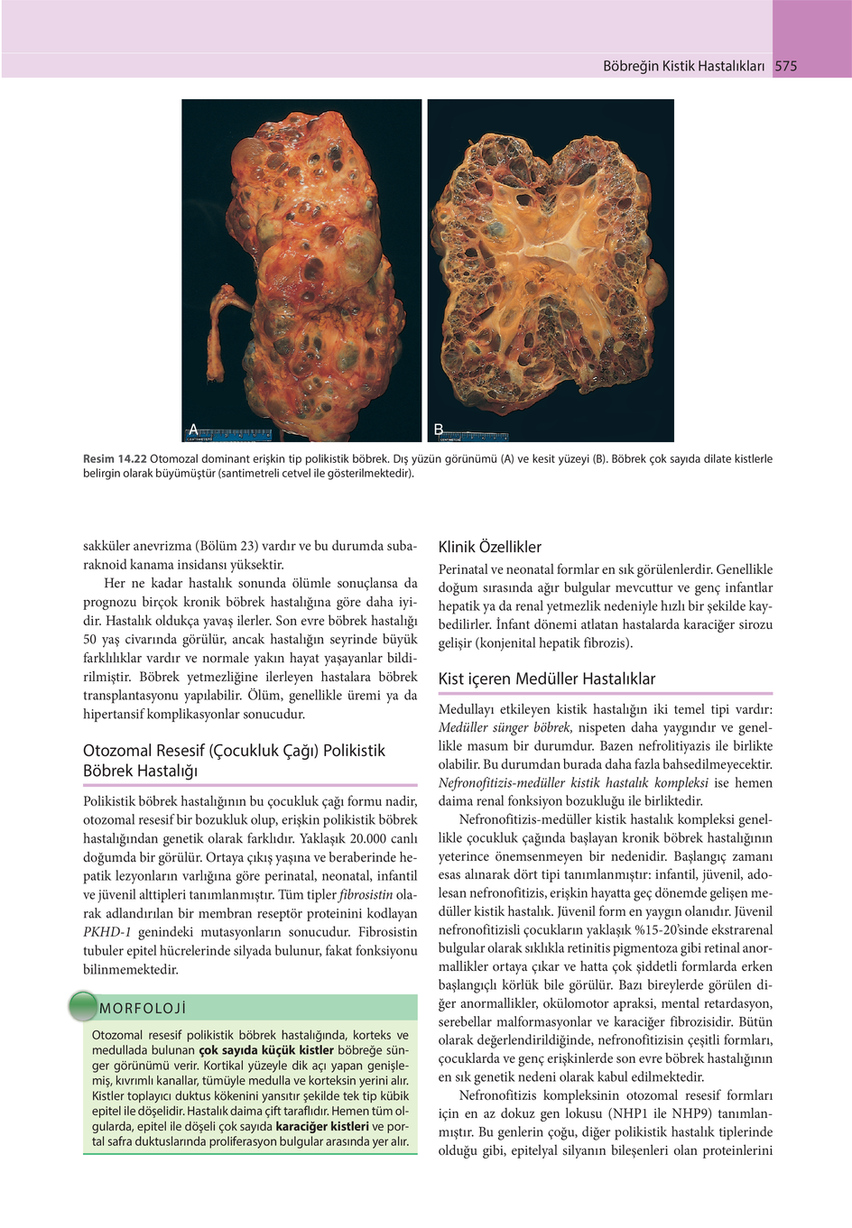
Resim 14.22 Otomozal dominant erişkin tip polikistik böbrek. Dış yüzün görünümü (A) ve kesit yüzeyi (B). Böbrek çok sayıda dilate kistlerle belirgin olarak büyümüştür (santimetreli cetvel ile gösterilmektedir).
sakküler anevrizma (Bölüm 23) vardır ve bu durumda subaraknoid kanama insidansı yüksektir. Her ne kadar hastalık sonunda ölümle sonuçlansa da prognozu birçok kronik böbrek hastalığına göre daha iyidir. Hastalık oldukça yavaş ilerler. Son evre böbrek hastalığı 50 yaş civarında görülür, ancak hastalığın seyrinde büyük farklılıklar vardır ve normale yakın hayat yaşayanlar bildirilmiştir. Böbrek yetmezliğine ilerleyen hastalara böbrek transplantasyonu yapılabilir. Ölüm, genellikle üremi ya da hipertansif komplikasyonlar sonucudur.
Klinik Özellikler
Perinatal ve neonatal formlar en sık görülenlerdir. Genellikle doğum sırasında ağır bulgular mevcuttur ve genç infantlar hepatik ya da renal yetmezlik nedeniyle hızlı bir şekilde kaybedilirler. İnfant dönemi atlatan hastalarda karaciğer sirozu gelişir (konjenital hepatik fibrozis).
Kist içeren Medüller Hastalıklar
Medullayı etkileyen kistik hastalığın iki temel tipi vardır: Medüller sünger böbrek, nispeten daha yaygındır ve genellikle masum bir durumdur. Bazen nefrolitiyazis ile birlikte olabilir. Bu durumdan burada daha fazla bahsedilmeyecektir. Nefronofitizis-medüller kistik hastalık kompleksi ise hemen daima renal fonksiyon bozukluğu ile birliktedir. Nefronofitizis-medüller kistik hastalık kompleksi genellikle çocukluk çağında başlayan kronik böbrek hastalığının yeterince önemsenmeyen bir nedenidir. Başlangıç zamanı esas alınarak dört tipi tanımlanmıştır: infantil, jüvenil, adolesan nefronofitizis, erişkin hayatta geç dönemde gelişen medüller kistik hastalık. Jüvenil form en yaygın olanıdır. Jüvenil nefronofitizisli çocukların yaklaşık %15-20’sinde ekstrarenal bulgular olarak sıklıkla retinitis pigmentoza gibi retinal anormallikler ortaya çıkar ve hatta çok şiddetli formlarda erken başlangıçlı körlük bile görülür. Bazı bireylerde görülen diğer anormallikler, okülomotor apraksi, mental retardasyon, serebellar malformasyonlar ve karaciğer fibrozisidir. Bütün olarak değerlendirildiğinde, nefronofitizisin çeşitli formları, çocuklarda ve genç erişkinlerde son evre böbrek hastalığının en sık genetik nedeni olarak kabul edilmektedir. Nefronofitizis kompleksinin otozomal resesif formları için en az dokuz gen lokusu (NHP1 ile NHP9) tanımlanmıştır. Bu genlerin çoğu, diğer polikistik hastalık tiplerinde olduğu gibi, epitelyal silyanın bileşenleri olan proteinlerini
Otozomal Resesif (Çocukluk Çağı) Polikistik Böbrek Hastalığı
Polikistik böbrek hastalığının bu çocukluk çağı formu nadir, otozomal resesif bir bozukluk olup, erişkin polikistik böbrek hastalığından genetik olarak farklıdır. Yaklaşık 20.000 canlı doğumda bir görülür. Ortaya çıkış yaşına ve beraberinde hepatik lezyonların varlığına göre perinatal, neonatal, infantil ve jüvenil alttipleri tanımlanmıştır. Tüm tipler fibrosistin olarak adlandırılan bir membran reseptör proteinini kodlayan PKHD-1 genindeki mutasyonların sonucudur. Fibrosistin tubuler epitel hücrelerinde silyada bulunur, fakat fonksiyonu bilinmemektedir.
M O R F O LO J İ
Otozomal resesif polikistik böbrek hastalığında, korteks ve medullada bulunan çok sayıda küçük kistler böbreğe sünger görünümü verir. Kortikal yüzeyle dik açı yapan genişlemiş, kıvrımlı kanallar, tümüyle medulla ve korteksin yerini alır. Kistler toplayıcı duktus kökenini yansıtır şekilde tek tip kübik epitel ile döşelidir. Hastalık daima çift taraflıdır. Hemen tüm olgularda, epitel ile döşeli çok sayıda karaciğer kistleri ve portal safra duktuslarında proliferasyon bulgular arasında yer alır.