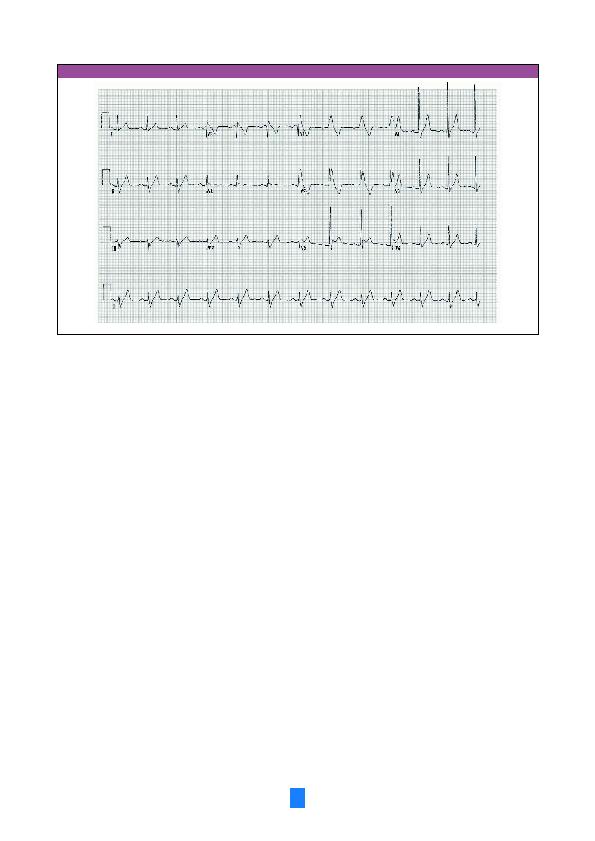
vertraagde intraventriculaire geleiding geeft een beeld van rechterbun-
deltakblok en een omkering van de T-top in de afleidingen V1-V3. In 50%
van de gevallen tonen de rechterafleidingen een epsilongolf (Figuur 5)
in de vorm van een `inkeping' op het einde van het QRS-complex (7). Die
epsilongolf is eveneens een teken van tragere intraventriculaire gelei-
ding. In verder gevorderde gevallen vertonen die patiënten een dilatatie
van het rechterventrikel bij echocardiografie, angiografie of magneti-
sche kernspinresonantie van het hart. Die afwijking kan het orificium
van de tricuspidalisklep openrekken met tricuspidalsinsufficiëntie als
gevolg en kan ook leiden tot een paradoxale beweging van het septum
en dilatatie van het uitstroomkanaal.
voor er macroscopische tekenen verschijnen en heeft dan ook een hoge
diagnostische waarde. De ventrikeltachycardie bestaat uit een macrore-
entrycircuit rond het geïnfiltreerde weefsel en kan leiden tot plotselinge
dood, vooral bij jonge sporters (8). Een elektrofysiologisch onderzoek is
alleen geïndiceerd om het risico op plotselinge dood te ramen. Genees-
middelen werken niet. Meestal wordt een defibrillator geplaatst.
ding in het ventrikel en dat laatste werkt maligne ritmestoornissen zoals
torsades de pointes in de hand. Een lang-QT-interval kan aangeboren
zijn of kan worden veroorzaakt door bepaalde geneesmiddelen, toxines,
catecholamineproducerende tumoren en elektrolytenstoornissen (Figu-
ren 6 en 7).
Lange-Nielsen, syndroom van Romano-Ward). De verlenging van het
QT-interval is toe te schrijven aan één of meer genetische afwijkingen.
We kennen momenteel drie belangrijke vormen: LQT 1 (afwijking van
het KvLQT1-gen op chromosoom 11), LQT 2 (afwijking van het HERG-gen
op chromosoom 7; dat gen heeft te maken met de kaliumkanalen) en
heeft met de natriumkanalen) (9).
syndroom worden veroorzaakt door vertraagde, vroege of late depolari-
saties (delayed after depolarizations) die de ventriculaire ritmestoornis
in gang zetten (10). De symptomen van een lang-QT-syndroom kunnen
beginnen op elke leeftijd met episoden van malaise, hartkloppingen,
syncope en plotselinge dood. De incidentie van torsades de pointes en
plotselinge dood door ventrikelfibrillatie is hoog. Zonder behandeling
bedraagt de sterfte 70% na 10-15 jaar (11).
terval gecorrigeerd voor de hartfrequentie (QTc). Bepaalde afwijkingen
van de T-golf en bepaalde klinische beelden correleren direct met een
genetische mutatie. Familiale screening is obligaat: bij alle biologische
familieleden van de betrokken patiënt moet een ecg worden afgeno-
men. Tot slot moet een genetisch onderzoek worden verricht met opspo-
ring van de belangrijkste 3 mutaties.
optreden van symptomen bij 80% van de jongeren (12). In geval van
recidiverende syncopes ondanks een behandeling met bètablokkers kan
een linker stellectomie worden overwogen, maar in refractaire gevallen
is inplanting van een automatische defibrillator de enige mogelijkheid.
wordt gekenmerkt door maligne ventriculaire ritmestoornissen (ventri-
keltachycardie, ventrikelfibrillatie, plotselinge dood) bij patiënten met
een gezond hart. Meestal beginnen de ritmestoornissen vanaf de leef-
tijd van 40 jaar, maar in de literatuur zijn enkele gevallen van plotselinge
dood beschreven bij kinderen met het brugadasyndroom. Het syndroom
wordt veroorzaakt door een mutatie van het SCN5a-gen van het natri-
umkanaal van de myocardcellen.